Automated Workflows in CryoSPARC™
How to make use of one-click, automated data processing workflows that can provide equal or better results as manual processing
Single particle cryo-EM is a powerful technique for life science research and drug discovery, with applications ranging from structure-based drug design, epitope mapping and time-resolved studies, to the characterization of disease-associated mutations, allosteric regulation and validation of synthetic biologic protein designs.
As the scope of structural biology continues to expand, the technique continues to grow in its reach and impact. However, cryo-EM data processing is still complex, generally requiring an expert scientist in the loop in order to make key experimental decisions to arrive at state of the art results, which limits the throughput of structure determination.
We recently set out to determine if there was a way to make cryo-EM data processing more routine, scalable, and reproducible. This challenge is particularly important in a context like structure-based drug design (SBDD), and especially in the “repeat-target" scenario (solving the same target structure with different ligands).
In this post, we explore the development of an automated data processing workflow that meets or exceeds the quality of manually-processed, human-driven results, even for the most challenging targets.
Automated workflow for GPCRs: developing a generalizable strategy for repeat-target data processing
In our recent preprint, we present the development of a concrete, end to end solution for automation of single particle analysis, demonstrated on 21 GPCR datasets and extensible to many other targets.
We demonstrate that automated data processing can achieve results equal or better than manual processing and that a single workflow can be generalized across diverse datasets and targets. To encourage adoption and experimentation, we make available the workflow JSON file and key considerations for automated processing, allowing users to immediately deploy, adapt and customize the pipeline to their own processing environment and research needs.
GPCRs as a real-world test case
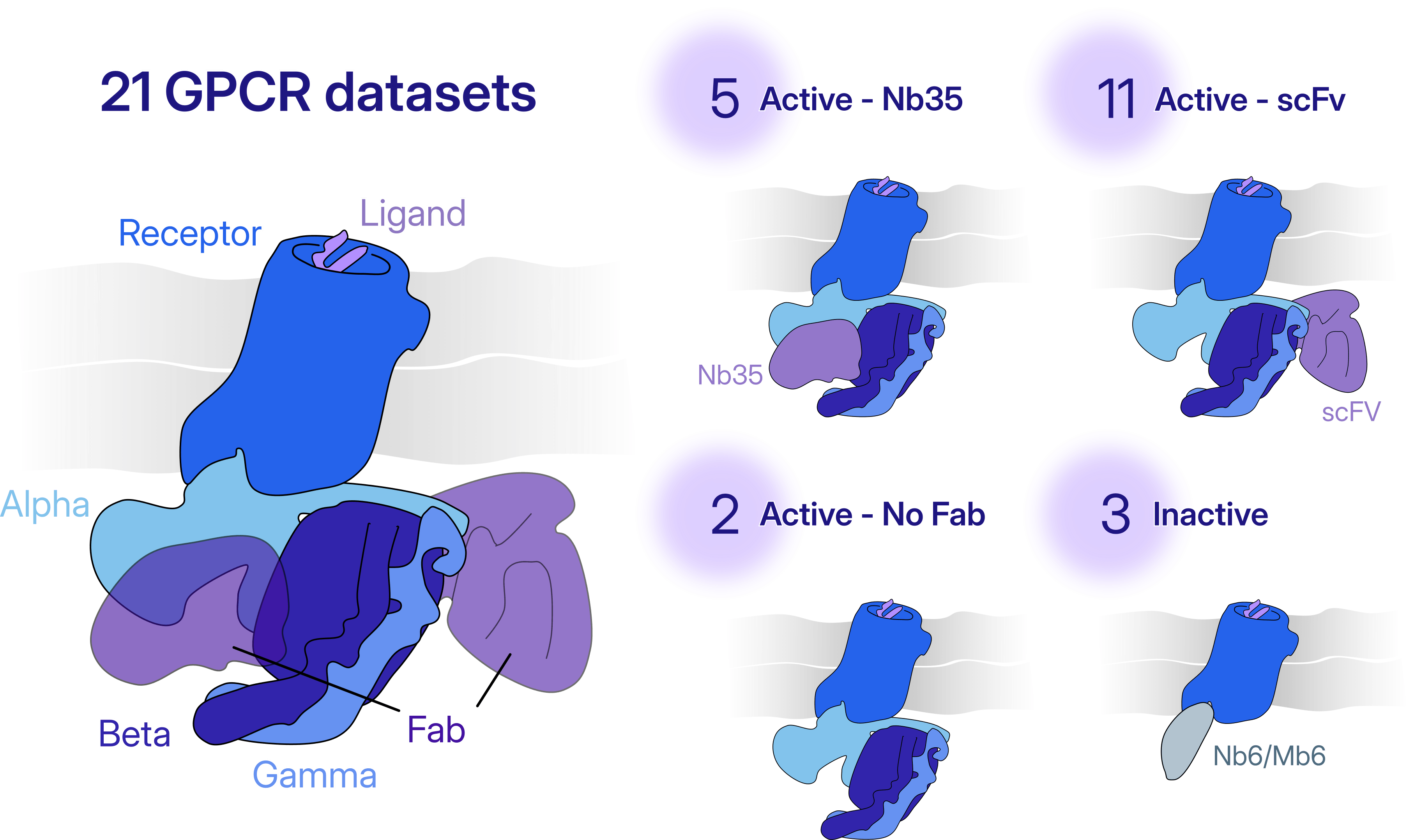
GPCRs represent a biologically and pharmaceutically important protein family targeted by approximately 36% of current FDA-approved drugs. We considered 21 publicly deposited GPCR datasets organized into four target classes according to their state (Active - unbound from Fab, Active - bound to scFV16/30, Active - bound to Nb35, and Inactive). Using our automated workflow, for 17 of the 21 datasets we were able to meet or exceed resolution and map quality compared to published results. For the other four datasets, we achieve resolutions within 0.2 Å and similar map quality compared to the published depositions.
Minimal Prior Information and Inputs
One of the key challenges in making automation possible was that only limited prior information about the target class should be required. Ultimately, through the development of an automated processing strategy and extensive experimentation, we arrived at a minimal set of inputs that a user would have to provide in order to use the workflow on any new dataset:
- Dataset level inputs: Microscope and acquisition parameters such as pixel size, acceleration voltage, spherical aberration, and total dose, which are readily available for each new dataset.
- Class level inputs: For each new target class (in our experiments, 21 GPCR datasets were classified into four target classes), a low-resolution reference map (~15 Å), random junk volumes, a mask defining the region of interest for local refinement (for example, a ligand-binding pocket), and the expected particle size and separation distance.
- Workflow level inputs: These remain constant across different targets, and they include parameters such as the number of decoy-classification rounds and the selection thresholds used for automated 2D and 3D class selection.
Results from automated processing of GPCRs
For the majority of GPCR datasets we tested, our automated workflow yielded higher-quality and more interpretable maps compared to manual processing. In addition, the map quality in the ligand-binding pocket across the various datasets was similar or improved. The automated workflow was able to robustly curate datasets consisting of millions of initial particles, even though the datasets varied by the species of GPCR and the amount of junk that was present. And most importantly, we were able to show that the workflow generalizes well between target classes, using only limited prior information, making it possible to extend to other targets.
Why did the automated workflow perform so well?
Building a fully automated cryo-EM workflow is not simply a matter of connecting existing processing steps together. At several stages of the pipeline, expert users routinely make decisions that determine whether processing succeeds or fails.
We systematically identified these bottlenecks and developed new methods that allow the workflow to work in practice. Several of these innovations proved critical to achieve high-quality reconstructions across diverse datasets.
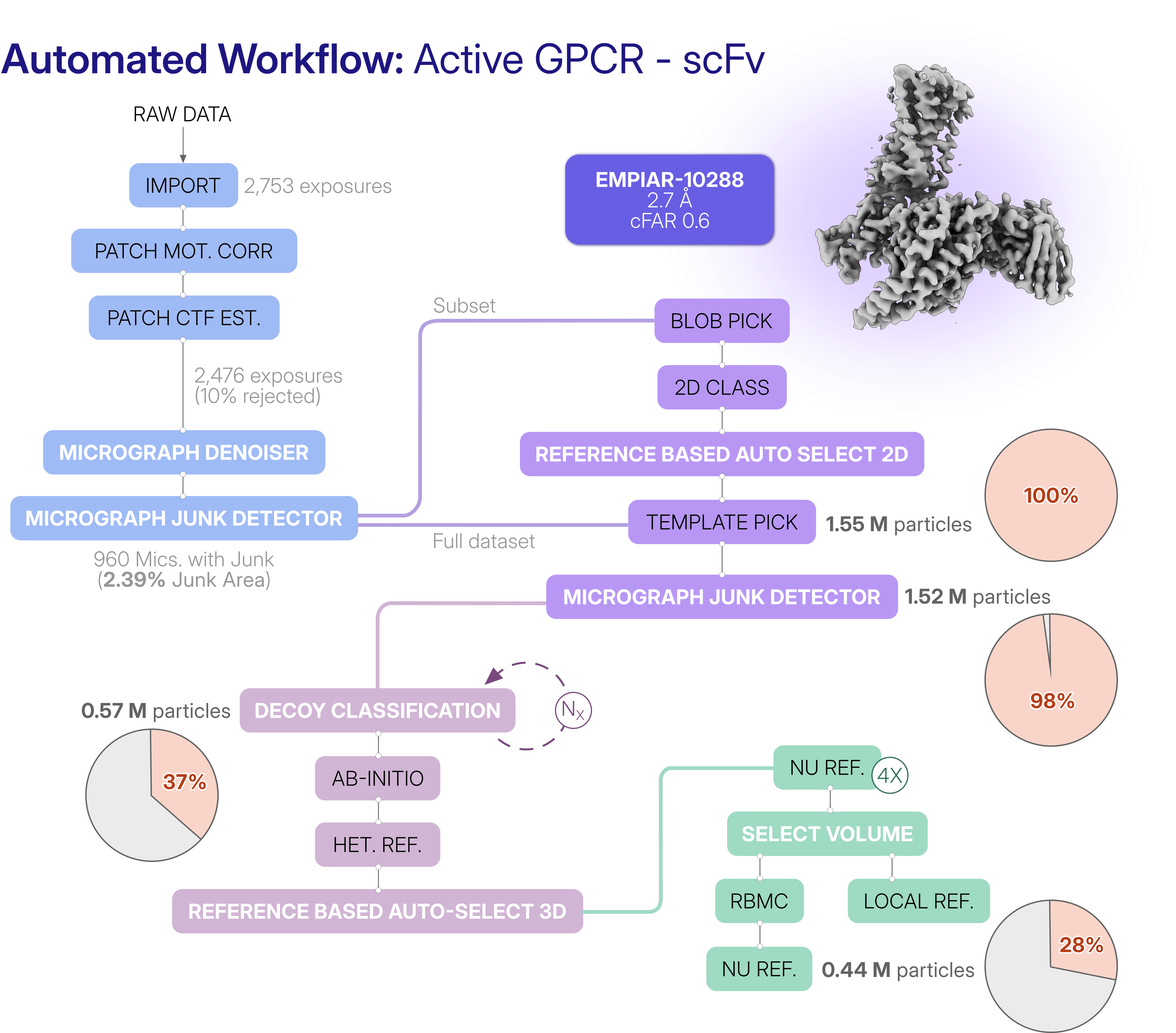
- Preprocessing: In a conventional workflow, users inspect micrographs and manually decide which ones (and which regions within these micrographs) are suitable for particle picking. To automate this decision-making process, we developed the Micrograph Denoiser, which improves image quality and enables reliable particle picking even in challenging datasets. We complemented this with Micrograph Junk Detector, which automatically excludes regions containing contaminants or damaged ice. Together, these methods allow the workflow to identify suitable picking regions without user intervention, providing a robust foundation for all subsequent processing steps.
- Particle Picking: Another challenge was automating the particle selection while retaining as many candidate picks as possible, without sacrificing rare views. Typically, a user would inspect and modify particle picks manually, adjusting thresholds in the interactive job. To automate this process, we developed Reference Based Auto Select 2D, which enables non-interactive 2D class selection based on a provided 3D reference volume. This job allows a broader and more representative selection of 2D classes, including rare views that might otherwise be discarded.
- Particle Curation: Even after particle picking, a major challenge remains: separating target particles from junk, while preserving rare views. In conventional cryo-EM processing, this step relies heavily on user-guided inspection of 2D and 3D classification results, making it difficult to automate reliably and reproducibly. To overcome this challenge, we employed a decoy classification strategy that uses a low-resolution reference volume and a set of junk volumes during Heterogeneous Refinement. All picked particles are automatically sorted into target and decoy classes, and only particles assigned to the target class are carried forward into multi-class Ab-Initio Reconstruction and downstream Heterogeneous Refinement. We then developed the Reference Based Auto Select 3D job, that automatically identifies the best quality classes.
- Refinement: Experienced users routinely inspect maps, compare refinement results, and decide which processing strategy yields the highest quality and most interpretable maps. This decision-making process is difficult to automate because good map quality is typically achieved through dataset-specific refinement strategies. To overcome this challenge, we designed the automated workflow to explore multiple refinement settings in parallel, allowing different parameter combinations to be evaluated simultaneously. We then developed the Select Volume job, which automatically identifies the best reconstruction. Local Refinement and Reference-Based Motion Correction are then applied to maximize map quality and resolution.
Adapting the GPCR workflow for other target classes
Our strategy can be applied to any type of target, including but not limited to, membrane proteins, soluble proteins, etc. The provided workflow JSON file can be readily adapted to different datasets by adjusting key parameters, including the targel level, class level and workflow level inputs described above. Together, these advances help increase the scalability and throughput of cryo-EM data processing, lowering the barrier for new and expert cryo-EM practitioners to obtain high-quality results capable of driving downstream biological and drug-discovery decisions.
While this work demonstrates that scalable and generalizable automated cryo-EM processing is already achievable, development at Structura Biotechnology continues toward workflows capable of tackling even more challenging problems, including structural heterogeneity and preferred orientation. Stay tuned for more updates!