A Complex Story: A Case Study in Cryo-EM Heterogeneity
From active and deactive states to additional respiratory complexes, this case study shows how a single cryo-EM dataset can tell many different stories when viewed through the lens of heterogeneity.
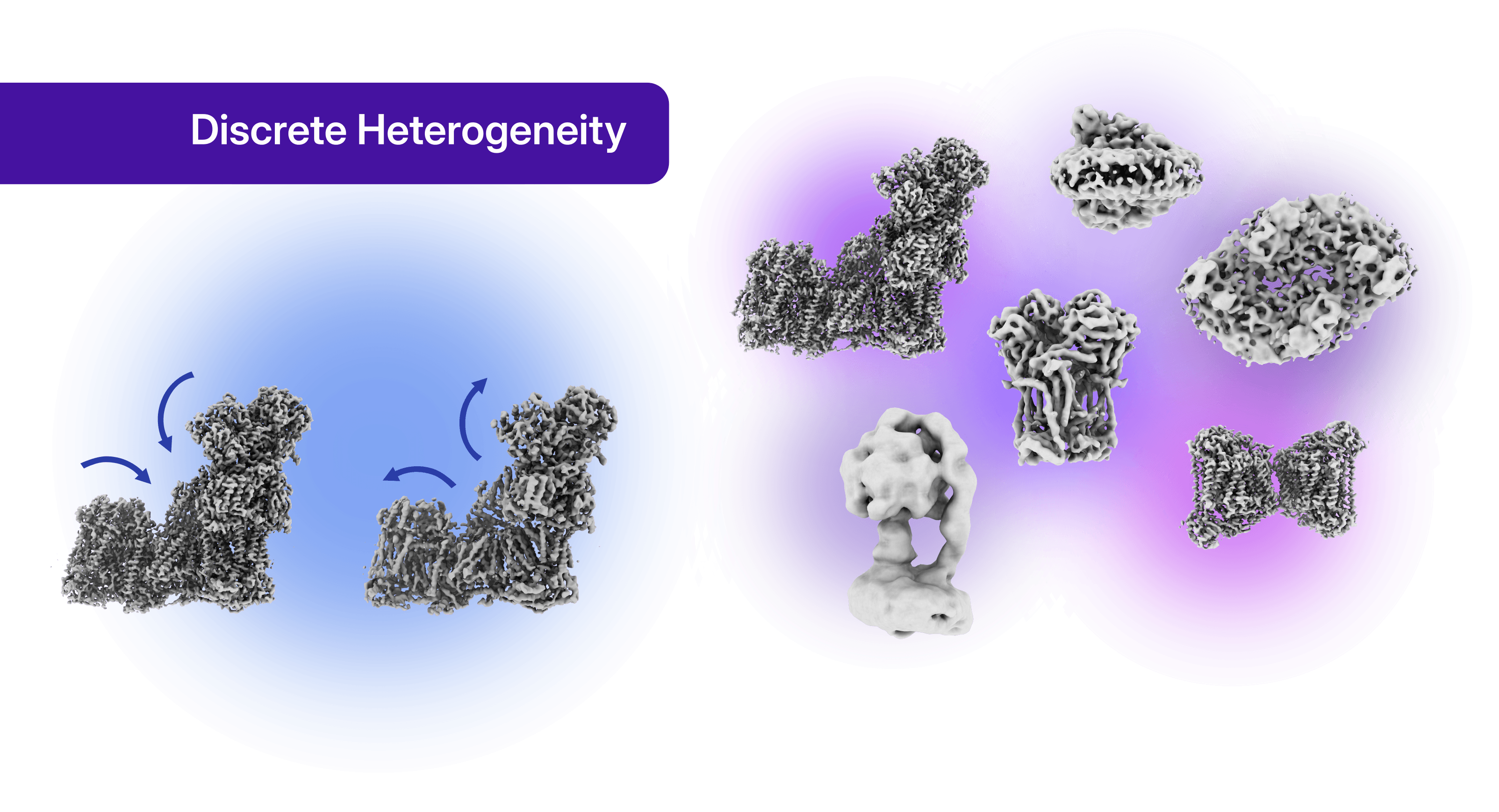
Cryo-EM offers a unique opportunity to explore biological complexity by separating and characterizing different molecular states present within a single sample. This heterogeneity can be broadly classified into two categories: continuous and discrete. Continuous heterogeneity describes the dynamic motions and conformational changes of proteins and macromolecular assemblies. Capturing these movements is often essential for understanding molecular mechanisms, revealing how biological machines function and transition between different functional states. Discrete heterogeneity arises from the presence of distinct molecular species or assemblies within the sample.
Identifying these populations can uncover transient interaction partners, neighboring membrane complexes, or different oligomeric states, all of which may be directly linked to biological function. Importantly, discrete heterogeneity can also provide the opportunity to determine multiple structures from the same dataset, substantially increasing the scientific value of a single experiment.
The dataset examined in the recently published Case Study: Acetogenin-bound Mitochondrial Complex I (EMPIAR 10927) consists of mouse heart mitochondrial respiratory complex I prepared in the presence of the tight-binding inhibitor acetogenin and processed using CryoSPARC v5. It provides an excellent example of how discrete heterogeneity can be explored through alternative data processing strategies.
This dataset has become a popular benchmark in cryo-EM software development because complex I is a large particle (1 MDa) and the dataset presents high signal-to-noise ratio, enabling high-resolution structure determination from a relatively modest dataset of only 1,283 micrographs. Conventional processing readily produces an interpretable reconstruction of complex I bound to acetogenin, deposited as PDB:7PSA. However, the true richness of this dataset emerges when different heterogeneity analysis strategies are applied.
Depending on the processing workflow, two well-characterized conformational states of complex I, commonly referred to as the active and deactive states (also known as the closed and open states, respectively) can be resolved. At the same time, discrete heterogeneity analysis reveals additional respiratory-chain complexes co-purified with complex I, including complexes III, IV, and V. Remarkably, these structures can be recovered from the same particle stack used to determine the complex I reconstruction in the first strategy presented in this case study.
This case study highlights several approaches for handling heterogeneity, illustrating how different processing strategies can be tailored to answer specific biological questions and maximize the structural information extracted from a single cryo-EM dataset.
Strategies to Detect and Handle Sample Heterogeneity
Particle Picking
The first opportunity to capture heterogeneity arises during particle picking. One effective strategy for this dataset was the use of TOPAZ, which differs from traditional picking approaches by not requiring a predefined particle diameter. Instead, TOPAZ relies on a minimum distance between particle centers, allowing particles of different sizes to be selected simultaneously. This can be particularly advantageous when exploring discrete heterogeneity, as it enables the recovery of multiple molecular species that may coexist within the sample.
 Knowledge or simply setting of particle diameter is not strictly necessary for TOPAZ picking, and the micrographs can be manually downsampled to a pixel size where particle features are still visible. Additionally the radius of extracted regions can be calculated from the interparticle distance, downsample factor and pixel size, as described in the case study.
Knowledge or simply setting of particle diameter is not strictly necessary for TOPAZ picking, and the micrographs can be manually downsampled to a pixel size where particle features are still visible. Additionally the radius of extracted regions can be calculated from the interparticle distance, downsample factor and pixel size, as described in the case study.
Per-Particle Scale
CryoSPARC v5 refinement jobs estimate per-particle scale factors by default. Examining the distribution of these scale values can provide valuable insight into the composition and quality of the particle stack. In some cases, multimodal distributions emerge, suggesting the presence of distinct particle populations. Since per-particle scaling affects the contribution of individual particles to the reconstructed volume when Minimize over per particle scale is set, separating particles according to their scale factors can help identify underlying compositional or conformational differences. These subsets can be generated using the Subset Particles by Statistics job and processed independently to further investigate their structural characteristics.
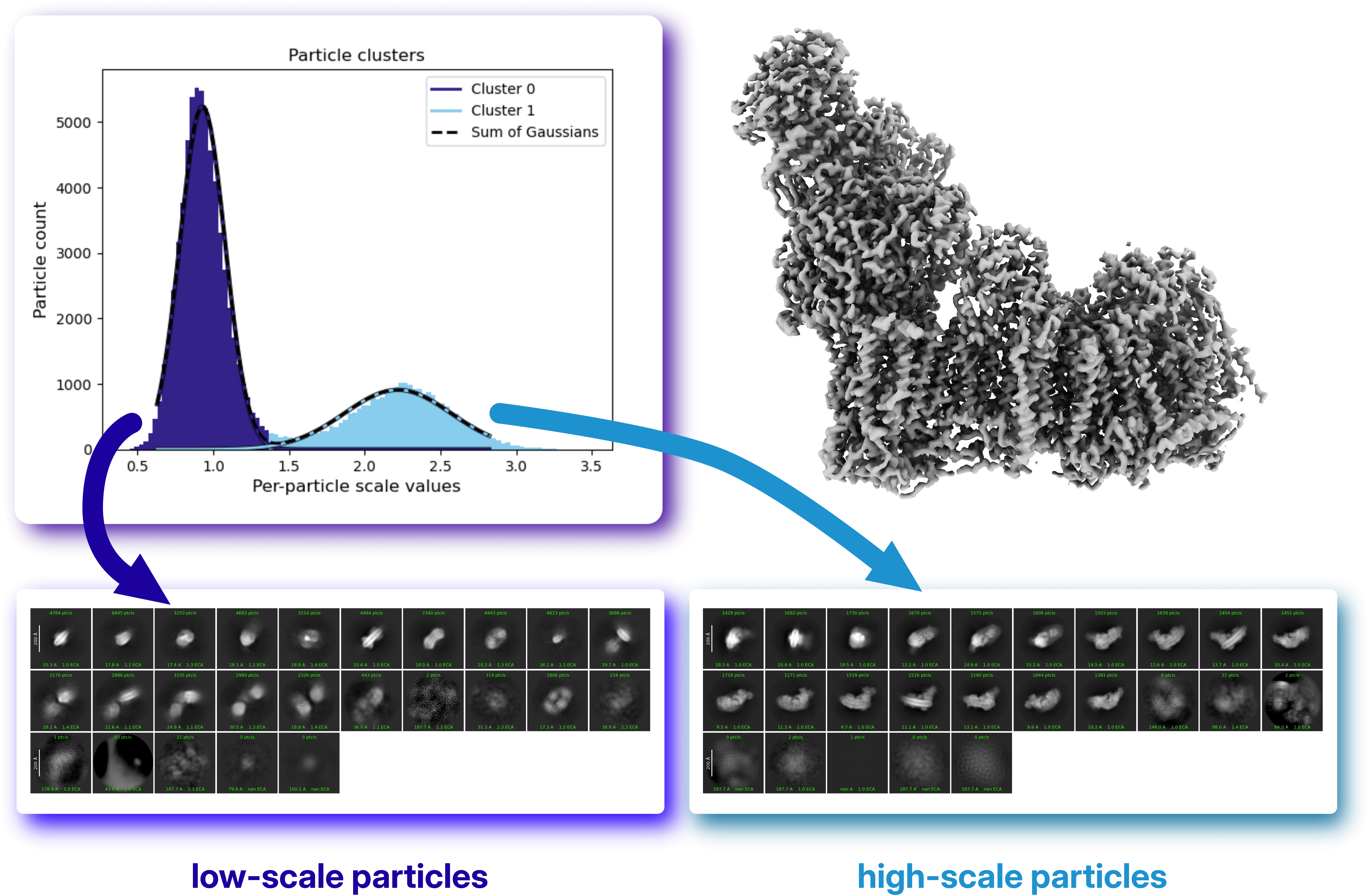 In this example from the case study, the particle stack used to reconstruct complex I exhibits a bimodal distribution of per-particle scale factors. Further analysis of the two particle populations reveals that the low-scale cluster corresponds largely to different molecular species present in the dataset. This result is particularly noteworthy because the structure was obtained using Non-Uniform Refinement with "minimize over per-particle scale" enabled, a setting that reduces the contribution of particles that do not match the reference volume. As a consequence, the per-particle scale distribution itself becomes a useful indicator of underlying sample heterogeneity.
In this example from the case study, the particle stack used to reconstruct complex I exhibits a bimodal distribution of per-particle scale factors. Further analysis of the two particle populations reveals that the low-scale cluster corresponds largely to different molecular species present in the dataset. This result is particularly noteworthy because the structure was obtained using Non-Uniform Refinement with "minimize over per-particle scale" enabled, a setting that reduces the contribution of particles that do not match the reference volume. As a consequence, the per-particle scale distribution itself becomes a useful indicator of underlying sample heterogeneity.Ab-Initio Reconstruction
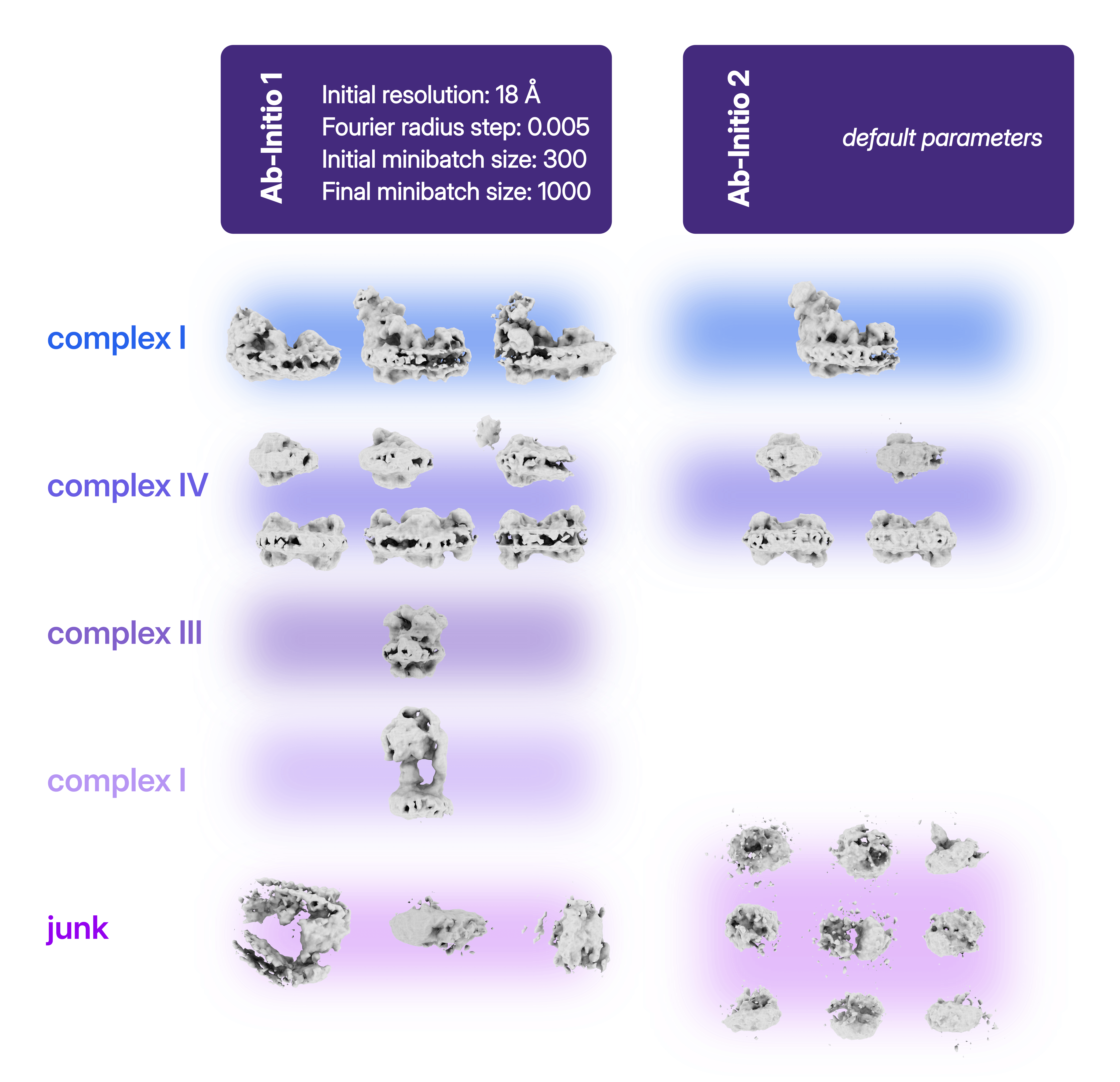
Ab-Initio Reconstruction offers a complementary approach by identifying structural populations without relying on an input reference volume. As discussed in our previous blog post on HR-HAIR and Homogeneous Ab-Initio Refinement, the parameters chosen for Ab-Initio Reconstruction strongly influence the amount of information that can be extracted from a dataset.
In the presented case study, the results of a standard Ab-Initio Reconstruction were compared with a medium-resolution Ab-Initio Reconstruction using an initial resolution of 18 Å and a reduced Fourier radius step of 0.005, the latter approach applied to the full particle stack. Running multi-class Ab-Initio Reconstruction with finer refinement steps can reveal distinct molecular species present in the dataset. The resulting classes can then serve as input references for Heterogeneous Refinement, allowing the separation between populations to be further improved.
3D Variability Analysis (3DVA)
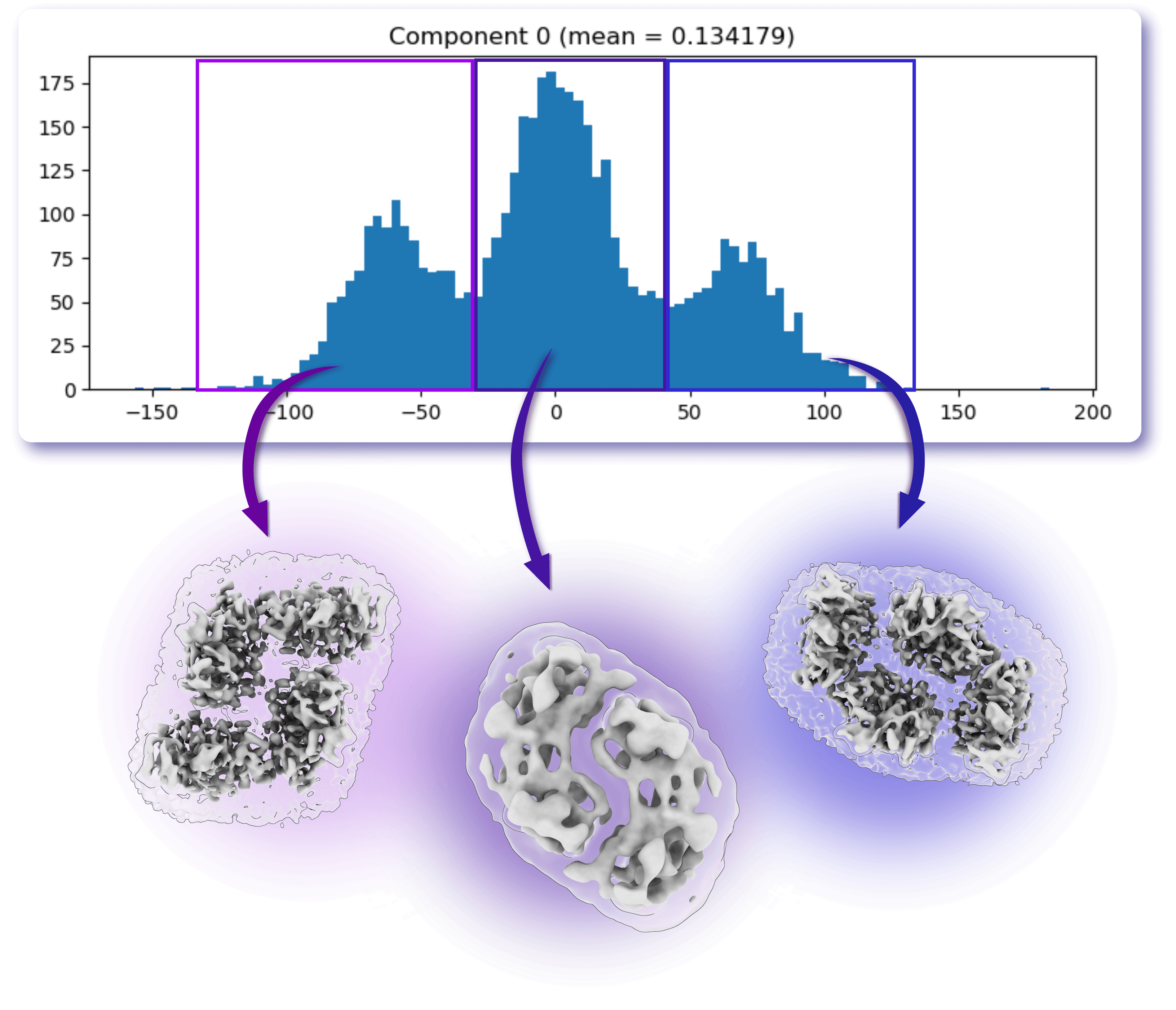
3D Variability Analysis (3DVA) is a powerful tool for exploring heterogeneity. During 3DVA, particle poses remain fixed according to their alignment to a reference refinement volume, while variability components are extracted and ranked according to the magnitude of the observed density changes. The resulting motions, visualized with 3D Variability Display, can reveal biologically relevant conformational transitions. However, interpretation requires caution, as some components may reflect reconstruction artifacts or compositional variability rather than genuine molecular motions. When carefully validated, 3DVA can provide valuable clues about both continuous and discrete heterogeneity within a dataset, hence being used as diagnostic as well as for separating particle sets.
3D Classification
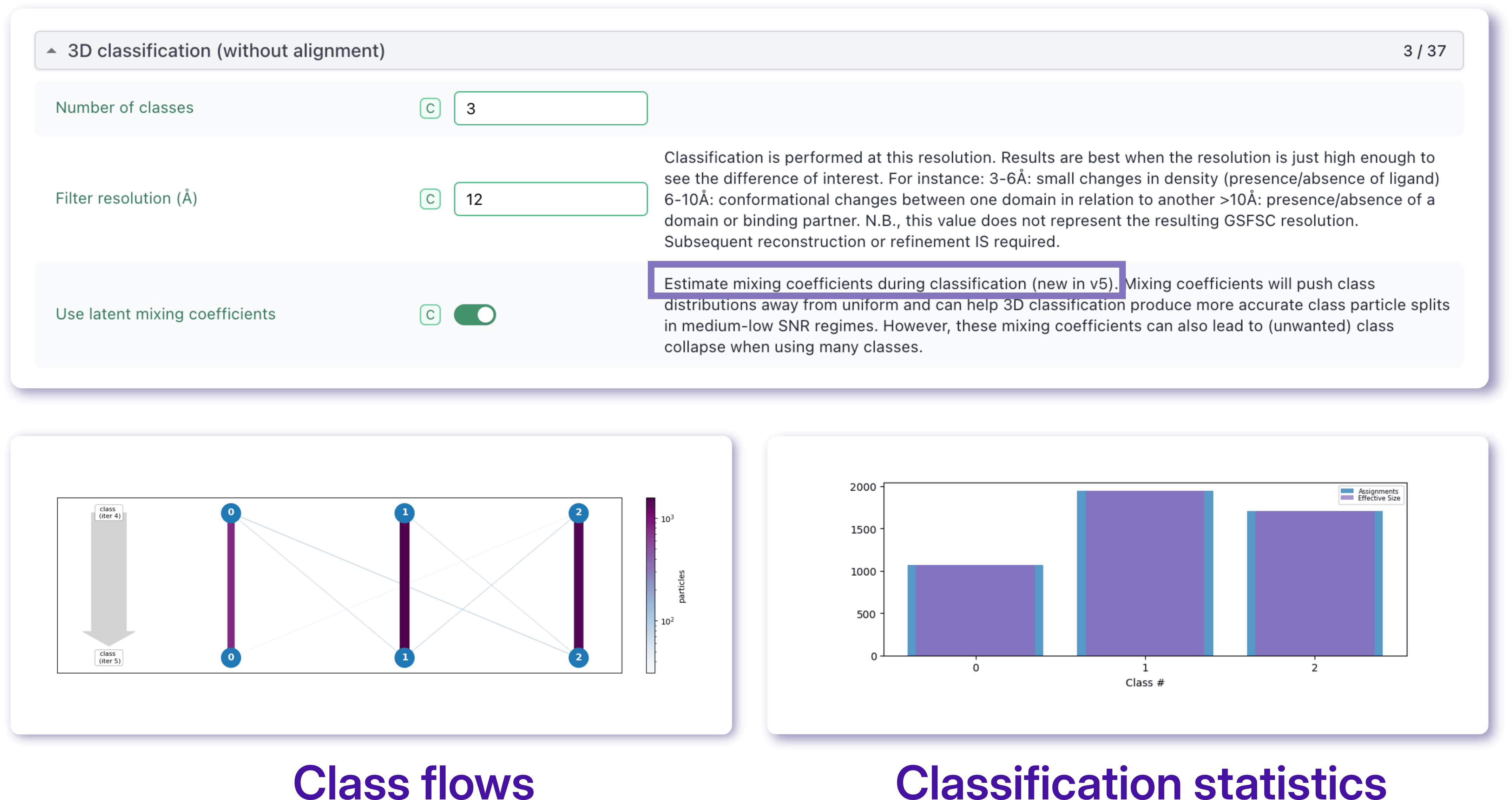
3D Classification remains one of the most direct approaches for identifying and separating distinct particle populations within a heterogeneous dataset. By exploring different parameter combinations, it is often possible to reveal conformational states, compositional variants, or low-abundance species that may remain hidden in a consensus reconstruction.
In CryoSPARC v5, 3D Classification introduces the option to enable latent mixing coefficients, providing a more flexible representation of particle assignments. This approach can improve class separation and reduce the tendency of the algorithm to generate multiple nearly identical classes containing similar particle counts. As a result, latent mixing coefficients can facilitate the identification of biologically meaningful populations and improve the recovery of rare or structurally distinct classes.
The Challenge of Class Separation
While numerous tools can reveal the presence of heterogeneity, the central challenge remains the same: effectively separating particles into meaningful classes. Conformational changes often exist alongside compositional differences, and the signatures of these phenomena can overlap. Successful analysis therefore can require combining multiple complementary strategies, iteratively refining particle assignments, always checking the resulting maps along side the job statistics, and adapting the workflow to the specific biological question being addressed.
The Case Study: Acetogenin-bound Mitochondrial Complex I (EMPIAR 10927) illustrates how a heterogeneous dataset can be analyzed using a combination of complementary strategies. While the workflow presented here proved effective for this particular sample, heterogeneity analysis is rarely a one-size-fits-all process, and the choice of methods should always be guided by the biological question of interest. For more inspiration and to learn more about the tools available to handle sample heterogeneity visit the CryoSPARC Guide and the CryoSPARC Discussion forum.